Какие функциональные группы содержатся в аминокислотах
Среди азотсодержащих органических веществ имеются соединения с двойственной функцией. Особенно важными из них являются аминокислоты.
В клетках и тканях живых организмов встречается около 300 различных аминокислот, но только 20 ( α-аминокислоты) из них служат звеньями (мономерами), из которых построены пептиды и белки всех организмов (поэтому их называют белковыми аминокислотами). Последовательность расположения этих аминокислот в белках закодирована в последовательности нуклеотидов соответствующих генов. Остальные аминокислоты встречаются как в виде свободных молекул, так и в связанном виде. Многие из аминокислот встречаются лишь в определенных организмах, а есть и такие, которые обнаруживаются только в одном из великого множества описанных организмов. Большинство микроорганизмов и растения синтезируют необходимые им аминокислоты; животные и человек не способны к образованию так называемых незаменимых аминокислот, получаемых с пищей. Аминокислоты участвуют в обмене белков и углеводов, в образовании важных для организмов соединений (например, пуриновых и пиримидиновых оснований, являющихся неотъемлемой частью нуклеиновых кислот), входят в состав гормонов, витаминов, алкалоидов, пигментов, токсинов, антибиотиков и т. д.; некоторые аминокислоты служат посредниками при передаче нервных импульсов.

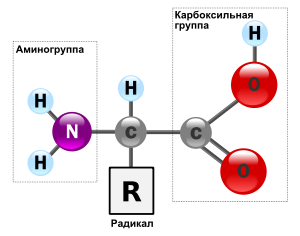
Аминокислоты можно рассматривать как карбоновые кислоты, в молекулах которых атом водорода в радикале замещен аминогруппой.
1. В зависимости от взаимного расположения амино- и карбоксильной групп аминокислоты подразделяют на α-, β-, γ-, δ-, ε- и т. д.

2. В зависимости от количества функциональных групп различают кислые, нейтральные и основные.
3. По характеру углеводородного радикала различают алифатические (жирные), ароматические, серосодержащие и гетероциклические аминокислоты. Приведенные выше аминокислоты относятся к жирному ряду.
Примером ароматической аминокислоты может служить пара-аминобензойная кислота:

Примером гетероциклической аминокислоты может служить триптофан – незаменимая α- аминокислота
По систематической номенклатуре названия аминокислот образуются из названий соответствующих кислот прибавлением приставки амино- и указанием места расположения аминогруппы по отношению к карбоксильной группе. Нумерация углеродной цепи с атома углерода карбоксильной группы.
Часто используется также другой способ построения названий аминокислот, согласно которому к тривиальному названию карбоновой кислоты добавляется приставка амино- с указанием положения аминогруппы буквой греческого алфавита.
Некоторые важнейшие α-аминокислоты
Наличие двух или трех карбоксильных групп отражается в названии суффиксом –диовая или -триовая кислота:

1. Изомерия углеродного скелета

2. Изомерия положения функциональных групп

3. Оптическая изомерия

Аминокислоты представляют собой кристаллические вещества с высокими (выше 250°С) температурами плавления, которые мало отличаются у индивидуальных аминокислот и поэтому нехарактерны. Плавление сопровождается разложением вещества. Аминокислоты хорошо растворимы в воде и нерастворимы в органических растворителях, чем они похожи на неорганические соединения. Многие аминокислоты обладают сладким вкусом.
Аминокислоты амфотерные органические соединения, для них характерны кислотно-основные свойства.
1. Внутримолекулярная нейтрализация → образуется биполярный цвиттер-ион:
Водные растворы электропроводны. Эти свойства объясняются тем, что молекулы аминокислот существуют в виде внутренних солей, которые образуются за счет переноса протона от карбоксила к аминогруппе:

Водные растворы аминокислот имеют нейтральную, кислую или щелочную среду в зависимости от количества функциональных групп.
Отношение аминокислот к индикаторам.mp4
2. Поликонденсация → образуются полипептиды (белки):
При взаимодействии двух α-аминокислот образуется дипептид.
3. Разложение → Амин + Углекислый газ:
II . Свойства карбоксильной группы (кислотность)
3. С аммиаком → образуются амиды:
4. Практическое значение имеет внутримолекулярное взаимодействие функциональных групп ε-аминокапроновой кислоты, в результате которого образуется ε-капролактам (полупродукт для получения капрона):

III . Свойства аминогруппы (основность)
1. С сильными кислотами → соли:
2. С азотистой кислотой (подобно первичным аминам):
Измерение объёма выделившегося азота позволяет определить количество аминокислоты (метод Ван-Слайка)
1. Все аминокислоты окисляются нингидрином с образованием продуктов сине-фиолетового цвета!
2. С ионами тяжелых металлов α-аминокислоты образуют внутрикомплексные соли. Комплексы меди (II), имеющие глубокую синюю окраску, используются для обнаружения α-аминокислот.

Образование медной соли аминоуксусной кислоты.mp4
1) аминокислоты широко распространены в природе;
2) молекулы аминокислот – это те кирпичики, из которых построены все растительные и животные белки; аминокислоты, необходимые для построения белков организма, человек и животные получают в составе белков пищи;
3) аминокислоты прописываются при сильном истощении, после тяжелых операций;
4) их используют для питания больных;
5) аминокислоты необходимы в качестве лечебного средства при некоторых болезнях (например, глутаминовая кислота используется при нервных заболеваниях, гистидин – при язве желудка);
6) некоторые аминокислоты применяются в сельском хозяйстве для подкормки животных, что положительно влияет на их рост;
7) имеют техническое значение: аминокапроновая и аминоэнантовая кислоты образуют синтетические волокна – капрон и энант.
Аминокислоты


Аминокисло́ты (аминокарбо́новые кисло́ты) — органические соединения, в молекуле которых одновременно содержатся карбоксильные и аминные группы.
Аминокислоты могут рассматриваться как производные карбоновых кислот, в которых один или несколько атомов водорода заменены на аминные группы.
Содержание
История
Открытие аминокислот в составе белков
| Аминокислота | аббревиатура | Год | Источник | Кто впервые выделил [1] |
|---|---|---|---|---|
| Глицин | Gly | 1820 | Желатин | А. Браконно |
| Лейцин | Leu | 1820 | Мышечные волокна | А. Браконно |
| Тирозин | Tyr | 1848 | Казеин | Ф. Бопп |
| Серин | Ser | 1865 | Шёлк | Э. Крамер |
| Глутаминовая кислота | Glu | 1866 | Растительные белки | Г. Риттхаузен |
| Аспарагиновая кислота | Asp | 1868 | Конглутин, легумин (ростки спаржи) | Г. Риттхаузен |
| Фенилаланин | Phe | 1881 | Ростки люпина | Э. Шульце, Й. Барбьери |
| Аланин | Ala | 1888 | Фиброин шелка | Т. Вейль |
| Лизин | Lys | 1889 | Казеин | Э. Дрексель |
| Аргинин | Arg | 1895 | Вещество рога | С. Гедин |
| Гистидин | His | 1896 | Стурин, гистоны | А. Кессель, С. Гедин |
| Цистеин | Cys | 1899 | Вещество рога | К. Мёрнер |
| Валин | Val | 1901 | Казеин | Э. Фишер |
| Пролин | Pro | 1901 | Казеин | Э. Фишер |
| Гидроксипролин | 1902 | Желатин | Э. Фишер | |
| Триптофан | Trp | 1902 | Казеин | Ф. Гопкинс, Д. Кол |
| Изолейцин | Ile | 1904 | Фибрин | Ф. Эрлих |
| Метионин | Met | 1922 | Казеин | Д. Мёллер |
| Треонин | Thr | 1925 | Белки овса | С. Шрайвер и др. |
| Гидроксилизин | 1925 | Белки рыб | С. Шрайвер и др. |
Физические свойства
Аминокислоты — бесцветные кристаллические вещества, хорошо растворимые в воде. Многие из них обладают сладким вкусом.
Общие химические свойства
Растворы аминокислот в воде благодаря этому обладают свойствами буферных растворов, т.е. находятся в состоянии внутренних солей.
Аминокислоты обычно могут вступать во все реакции, характерные для карбоновых кислот и аминов.
Важной особенностью аминокислот является их способность к поликонденсации, приводящей к образованию полиамидов, в том числе пептидов, белков, нейлона, капрона.
Изоэлектрической точкой аминокислоты называют значение pH, при котором максимальная доля молекул аминокислоты обладает нулевым зарядом. При таком pH аминокислота наименее подвижна в электрическом поле, и данное свойство можно использовать для разделения аминокислот, а также белков и пептидов.
Некоторые аминокислоты имеют несколько аминогрупп и карбоксильных групп. Для этих аминокислот трудно говорить о каком-то конкретном цвиттер-ионе.
Получение
Большинство аминокислот можно получить в ходе гидролиза белков или как результат химических реакций:
Оптическая изомерия
Все входящие в состав живых организмов α-аминокислоты, кроме глицина, содержат асимметричный атом углерода (треонин и изолейцин содержат два асимметричных атома) и обладают оптической активностью. Почти все встречающиеся в природе α-аминокислоты имеют L-форму, и лишь L-аминокислоты включаются в состав белков, синтезируемых на рибосомах.
Данную особенность «живых» аминокислот весьма трудно объяснить, так как в реакциях между оптически неактивными веществами L и D-формы образуются в одинаковых количествах. Возможно, выбор одной из форм (L или D) — просто результат случайного стечения обстоятельств: первые молекулы, с которых смог начаться матричный синтез, обладали определенной формой, и именно к ним «приспособились» соответствующие ферменты.
D-аминокислоты в живых организмах
Аспарагиновые остатки в метаболически неактивных структурных белках претерпевают медленную самопроизвольную неферментативную рацемизацию: так в белках дентина и эмали зубов L-аспартат переходит в D-форму со скоростью
С развитием следового аминокислотного анализа D-аминокислоты были обнаружены сначала в составе клеточных стенок некоторых бактерий (1966), а затем и в тканях высших организмов. Так, D-аспартат и D-метионин предположительно являются нейромедиаторами у млекопитающих.
В состав некоторых пептидов входят D-аминокислоты, образующиеся при посттрансляционной модификации. Например, D-метионин и D-аланин входят в состав опиоидных гептапептидов кожи южноамериканских амфибий филломедуз (дерморфина, дермэнкефалина и делторфинов). Наличие D-аминокислот определяет высокую биологическую активность этих пептидов как анальгетиков.
Сходным образом образуются пептидные антибиотики бактериального происхождения, действующие против грамположительных бактерий — низин, субтилин и эпидермин.
Гораздо чаще D-аминокислоты входят в состав пептидов и их производных, образующихся путем нерибосомного синтеза в клетках грибов и бактерий. Видимо, в этом случае исходным материалом для синтеза служат также L-аминокислоты, которые изомеризуются одной из субъединиц ферментного комплекса, осуществляющего синтез пептида.
Протеиногенные аминокислоты
В процессе биосинтеза белка в полипептидную цепь включаются 20 α-аминокислот, кодируемых генетическим кодом. Помимо этих аминокислот, называемых протеиногенными, или стандартными, в некоторых белках присутствуют специфические нестандартные аминокислоты, возникающие из стандартных в процессе посттрансляционных модификаций. В последнее время к протеиногенным аминокислотам иногда причисляют трансляционно включаемые селеноцистеин (Sec, U) и пирролизин (Pyl, O). Это так называемые 21-я и 22-я аминокислоты.
Вопрос, почему именно эти 20 аминокислот стали «избранными», остаётся не решённым. Не совсем ясно, чем эти аминокислоты оказались предпочтительнее других похожих. Например, ключевым промежуточным метаболитом пути биосинтеза треонина, изолейцина и метионина является α-аминокислота гомосерин. Очевидно, что гомосерин — очень древний метаболит, но для треонина, изолейцина и метионина существуют аминоацил-тРНК-синтетазы, тРНК, а для гомосерина — нет.
Структурные формулы 20-ти протеиногенных аминокислот обычно приводят в виде так называемой таблицы протеиногенных аминокислот:

Для запоминания однобуквенного обозначения протеиногенных аминокислот используется мнемоническое правило (последний столбец).
| Глицин | Gly | G | Glycine | Гли |
| Аланин | Ala | A | Alanine | Ала |
| Валин | Val | V | Valine | Вал |
| Изолейцин | Ile | I | Isoleucine | Иле |
| Лейцин | Leu | L | Leucine | Лей |
| Пролин | Pro | P | Proline | Про |
| Серин | Ser | S | Serine | Сер |
| Треонин | Thr | T | Threonine | Тре |
| Цистеин | Cys | C | Cysteine | Цис |
| Метионин | Met | M | Methionine | Мет |
| Аспарагиновая кислота | Asp | D | asparDic acid | Асп |
| Аспарагин | Asn | N | asparagiNe | Асн |
| Глутаминовая кислота | Glu | E | gluEtamic acid | Глу |
| Глутамин | Gln | Q | Q-tamine | Глн |
| Лизин | Lys | K | before L | Лиз |
| Аргинин | Arg | R | aRginine | Арг |
| Гистидин | His | H | Histidine | Гис |
| Фенилаланин | Phe | F | Fenylalanine | Фен |
| Тирозин | Tyr | Y | tYrosine | Тир |
| Триптофан | Trp | W | tWo rings | Три |
Классификация
По радикалу
По функциональным группам
По классам аминоацил-тРНК-синтетаз
Для аминокислоты лизин существуют аминоацил-тРНК-синтетазы обоих классов.
По путям биосинтеза
Пути биосинтеза протеиногенных аминокислот разноплановы. Одна и та же аминокислота может образовываться разными путями. К тому же совершенно различные пути могут иметь очень похожие этапы. Тем не менее, имеют место и оправданы попытки классифицировать аминокислоты по путям их биосинтеза. Существует представление о следующих биосинтетических семействах аминокислот: аспартата, глутамата, серина, пирувата и пентоз. Не всегда конкретную аминокислоту можно однозначно отнести к определённому семейству; делаются поправки для конкретных организмов и учитывая преобладающий путь. По семействам аминокислоты обычно распределяют следующим образом:
Фенилаланин, тирозин, триптофан иногда выделяют в семейство шикимата.
По способности организма синтезировать из предшественников
Классификация аминокислот на заменимые и незаменимые не лишена недостатков. К примеру, тирозин является заменимой аминокислотой только при условии достаточного поступления фенилаланина. Для больных фенилкетонурией тирозин становится незаменимой аминокислотой. Аргинин синтезируется в организме человека и считается заменимой аминокислотой, но в связи с некоторыми особенностями его метаболизма при определённых физиологических состояниях организма может быть приравнен к незаменимым. Гистидин также синтезируется в организме человека, но не всегда в достаточных количествах, потому должен поступать с пищей.
По характеру катаболизма у животных
Биодеградация аминокислот может идти разными путями. По характеру продуктов катаболизма у животных протеиногенные аминокислоты делят на три группы: глюкогенные (при распаде дают метаболиты, не повышающие уровень кетоновых тел, способные относительно легко становиться субстратом для глюконеогенеза: пируват, α-кетоглутарат, сукцинил-KoA, фумарат, оксалоацетат), кетогенные (распадаются до ацетил-KoA и ацетоацетил-KoA, повышающие уровень кетоновых тел в крови животных и человека и преобразующиеся в первую очередь в липиды), глюко-кетогенные (при распаде образуются метаболиты обоих типов).
«Миллеровские» аминокислоты
«Миллеровские» аминокислоты — обобщенное название аминокислот, получающихся в условиях, близких к эксперименту Стенли Л. Миллера 1953 года. Установлено образование в виде рацемата множества различных аминокислот, в том числе: глицин, аланин, валин, изолейцин, лейцин, пролин, серин, треонин, аспартат, глутамат
Родственные соединения
В медицине ряд веществ, способных выполнять некоторые биологические функции аминокислот, также (хотя и не совсем верно) называют аминокислотами:
Применение
Важной особенностью аминокислот является их способность к поликонденсации, приводящей к образованию полиамидов, в том числе пептидов, белков, нейлона, капрона, энанта.
Примечания
См. также
Ссылки
Miller S. L. Production of amino acids under possible primitive earth conditions. Science, v. 117, May 15, 1953
Miller S. L. and H. C. Urey. Organic compound synthesis on the primitive earth. Science, v. 130, July 31, 1959
Miller Stanley L. and Leslie E. Orgel. The origins of life on the earth. Englewood Cliffs, NJ, Prentice-Hall, 1974.
Лекция № 26 аминокислоты

А М И Н О К И С Л О Т Ы
Аминокислотами называют бифункциональные производные углеводородов, которые содержат карбоксильную группу —COOH и аминогруппу —NH2.
По систематической номенклатуре аминокислоты называют, по соответствующей карбоновой кислоте добавляя приставку амино-. Положение аминогруппы в углеродной цепи указывают цифрой:

В зависимости от положения аминогруппы по отношению к карбоксильной группе различают α, β, γ и так далее аминокислоты:

 Все природные аминокислоты содержат аминогруппу только в
Все природные аминокислоты содержат аминогруппу только в
α-положении и имеют общую формулу:
В настоящее время единой классификации аминокислот не существует.
Аминокислоты делят на природные (содержатся в растительных и животных организмах) и синтетические – получены искусственным путем.
Организм синтезирует аминокислоты главным образом из пищевых белков. Но есть целая группа аминокислот, которых организм сам синтезировать не может. Эти аминокислоты называют незаменимыми. К ним относятся (валин, лейцин, изолейцин, лизин, треонин, метионин, фенилаланин и триптофан) Такие аминокислоты должны поступать в организм извне.
В настоящее время известно свыше 150 аминокислот, но только 20 из них входят в состав белков.
По природе радикала аминокислоты делят на:
Строение радикала кислоты



Строение радикала кислоты



Строение радикала кислоты


4. Аминокислоты, содержащие в радикале дополнительную аминогруппу или гуанидильный остаток.
Строение радикала кислоты


Аргинин (содержит гунидиновую группу)
5. Аминокислоты, которые содержат в радикале дополнительную карбоксильную или амидную группы:
Строение радикала кислоты




6. Ароматические и гетероциклические аминокислоты:
Строение радикала кислоты




Пролин (полная форма)



Без участия ферментов самопроизвольный переход L-изомеров в D-изомеры с образованием эквимолярной смеси (рацемическая смесь) осуществляется в течение достаточно длительного промежутка времени.
Рацемизация каждой L-кислоты при данной температуре идет с определенной скоростью. Это обстоятельство можно использовать для установления возраста людей и животных. Так, например, в твердой эмали зубов имеется белок дентин, в котором L-аспартат переходит в D-изомер при температуре тела человека со скоростью 0,01% в год. В период формирования зубов в дентине содержится только L-изомер, поэтому по содержанию D-аспартата можно рассчитать возраст человека или животного.
Физические свойства аминокислот
Во-первых, в противоположность аминам и карбоновым кислотам аминокислоты представляют собой нелетучие кристаллические вещества, плавящиеся с разложением при близких и довольно высоких температурах, поэтому идентификации аминокислот по температурам плавления достаточно затруднительна.
Во-вторых, аминокислоты очень плохо растворимы в неполярных растворителях типа петролейного эфира, диэтилового эфира, бензола и хорошо растворимы в воде.
В-третьих, в водных растворах аминокислоты имеют высокие дипольные моменты.
В-четвертых, константы кислотности и основности для групп СООН и NH2 необычайно малы. Так, для глицина константа кислотности Ka = 1,6×10-10, а константа основности Kb = 2,5×10-12; в то время как для большинства карбоновых кислот Ka » 10-5 а для алифатических аминов Kb » 10-4. Все эти свойства вполне объяснимы, если принять во внимание тот факт, что аминокислоты существуют в виде диполярного иона, который образуется за счет отщепления протона от карбоксильной группы и присоединения его к аминогруппе. Диполярный ион часто называют внутренней солью.

Кислотно-основные свойства также становятся понятными, если учесть, что измеряемая Ka в действительности относится к кислотности иона RNH3+:

а константа основности (Kb) в действительности относится к основности карбоксилат-иона.


Если подкислить раствор аминокислоты, ион I превратится в катион III, так как более сильная кислота Н3О+ отдает протон карбоксилат-иону и образуется более слабая кислота:

Необходимо отметить, что ионы II и Ш, содержащие свободную аминогруппу или свободную карбоксильную группу, находятся в равновесии с диполярным ионом:

Однако следует иметь в виду, что в данном равновесии участвует также определенное (хотя и небольшое) количество незаряженных молекул аминокислот.
Изоэлектрическая точка аминокислот
Мы рассмотрели превращение в кислой и щелочной средах моноаминомонокарбоновых кислот, в радикалах которых не содержится ионогенных групп (аминокислоты с недиссоциирующими радикалами).
Изменение суммарного заряда аминокислот с анионными и катионными группами в радикале, в зависимости от рН среды, можно представить в следующей таблице. Для сравнения в эту же таблицу поместим аминокислоты, в радикале которых нет диссоциирующих групп.
Концентрация ионов водорода (pH), при которой аминокислота не перемещается в электрическом поле, называется изоэлектрической точкой данной аминокислоты (рI).
При пропускании постоянного тока через раствор, содержащий смесь нескольких аминокислот, каждая из них будет двигаться к катоду или к аноду со скоростью, зависящей от природы этой аминокислоты и от рН среды. Разделение и анализ смесей аминокислот, основанное на этом явлении, называется электрофорезом.
Химические свойства аминокислот
Наличие в молекуле аминокислоты функциональных групп кислотного и основного характера обусловливает амфотерность аминокислот. Подобно любому амфотерному соединению, аминокислоты образуют соли как при действии кислоты, так и при действии щелочи.

Аминокислоты, будучи гетерофункциональными соединениями, должны проявлять свойства как одной, так и другой функциональной группы.
Реакции карбоксильной группы
1. Образование внутрикомплексных солей.
С катионами тяжелых металлов α-аминокислоты образуют внутрикомплексные соли. Так, со свежеприготовленным гидроксидом меди (II) α-аминокислоты образуют хорошо кристаллизующиеся хелатные соли меди (II), окрашенные в синий цвет:

2. Образование сложных эфиров.
Так как реакция этерификации протекает в кислой среде, сложные эфиры аминокислот образуются в виде солей по аминогруппе:

Образовавшиеся эфиры не могут существовать в виде биполярных ионов, поэтому, в отличие от исходных аминокислот, они растворяются в органических растворителях и имеют более низкие температуры кипения. Это даёт возможность разделить смесь эфиров аминокислот перегонкой.
3. Образование хлорангидридов.
Эту реакцию часто называют реакцией «активации» карбоксильной группы. Хлорангидриды α-аминокислот получают действием на аминокислоты тионилхлорида (SOCl2) или хлорида фосфора (V) (PCl5). Полученные хлорангидриды неустойчивы и существуют только в виде солей:

Поэтому реакцию обычно проводят, предварительно защитив аминогруппу ацилированием.
4. Образование амидов аминокислот.
Такие амиды получают действием аммиака или первичных аминов на хлорангидриды с защищённой аминогруппой. В случае использования реакции с аминами получают замещённые по азоту амиды аминокислот:

5. Декарбоксилирование аминокислот.
В лабораторных условиях эта реакция протекает при нагревании аминокислоты с Ba(OH)2. В результате получается первичный амин:

Все реакции карбоксильной группы аминокислот можно представить следующей схемой:

1. Реакция ацилирования. Образование N-замещённых амидов.
N-замещенные амиды часто рассматривают как N-ацильные производные. Эта реакция была отмечена ранее как реакция защиты аминогруппы. Её можно рассматривать как процесс ацилирования аминогруппы хлорангидридами или ангидридами кислот:

Реакция протекает лучше в щелочной среде. Примером может служить получение N-бензоилаланина в присутствии водного раствора гидроксида натрия. Этот метод получения N-ацильных производных называют ацилированием по Шоттен-Бауману:

Щёлочь необходима для связывания выделяющегося хлороводорода, т. к. в кислой среде N-ацильные производные легко гидролизуются, освобождая исходную аминокислоту:

Это общепринятый способ удаления защитной группы. Однако в некоторых случаях невозможно удалять защитную группу гидролизом в кислой среде. Например, при гидролизе пептидов будет разрушаться пептидная связь. В этих случаях защиту проводят такими реагентами, удаление которых можно провести не гидролизом, а каким-либо другим методом. Например, аминогруппу можно защищать реакцией с карбобензоксихлоридом (бензиловый эфир хлормуравьиной кислоты). Карбобензоксигруппа удаляется затем каталитическим гидрогенолизом:

2. Алкилирование аминокислот.

При избытке иодистого метила образуется четвертичная аммонийная соль:


Таким образом можно установить структурное родство аминокислот с соответствующими оксикислотами. По объёму выделившегося азота определяют количество α-аминокислоты, вступившей в реакцию (метод Ван-Слайка).
4. Взаимодействие с альдегидами.
α-Аминокислоты, подобно первичным аминам, реагируют с альдегидами, образуя замещенные имины (основания Шиффа). Реакция протекает через стадию образования карбиноламинов.

Формальдегид, взятый в избытке, способствует отщеплению протона от NH3+ группы биполярного иона и легко соединяется со свободной (непротонированной) аминогруппой, образуя устойчивое метилольное производное.

Титрование аминокислоты в избытке формальдегида (формольное титрование) представляет собой аналитический метод (метод Серенсена), при помощи которого прослеживается, в частности, образование свободных аминокислот в процессе гидролиза белков.
5. Взаимодейстивие с динитрофторбензолом (ДНФБ).
Важной реакцией α-аминогруппы является её реакция с
2,4-динитрофторбензолом (ДНФБ) в слабощелочном растворе, которую впервые использовал Фредерик Сенгер для количественного введения метки в аминогруппы аминокислот и пептидов. Эта реакция протекает по механизму нуклеофильного замещения.

Продукт реакции окрашен в интенсивно желтый цвет. Эта реакция представляет исключительную ценность для идентификации N-концевых аминокислот полипептидных цепей.
Все вышеперечисленные реакции аминогруппы аминокислот можно представить следующей схемой:

Реакции функциональных групп, содержащихся в радикалах аминокислот
Аминокислоты вступают также в реакции, типичные для функциональных групп, присутствующих в их радикалах. Например, для SH-групп цистеина, гидроксильной группы тирозина и треонина, гуанидиновой группы аргинина.
1. Реакции сульфгидрильной (тиоловой) группы.
Для сульфгидрильной группы характерна исключительно высокая реакционная способность. Например, при действии на цистеин незначительных концентраций ионов некоторых тяжелых металлов образуются меркаптиды.

В щелочных растворах цистеин легко теряет атом серы. Так, при нагревании цистеина с ацетатом свинца в щелочном растворе образуется черный осадок сульфида свинца. Эта реакция применяется для обнаружения сульфгидрильной группы в пептидах и белках.
Тиоловая группа цистеина легко подвергается окислению с образованием дисульфида. Этот процесс можно отразить следующей схемой:

Дисульфидные связи, присоединяя два атома водорода, переходят в сульфгидрильные (тиоловые) группы:

Рассмотрим этот процесс на примере превращения цистеина в цистин:

В цистине при действии восстановителей дисульфидная связь разрывается и образуется две молекулы цистеина:

Дисульфидная связь может также подвергаться окислению под действием таких жестких окислителей, как, например, надмуравьиная кислота. В результате образуется цистеиновая кислота:

2. Реакции гидроксильной группы – реакции элиминирования.
Эти реакции характерны для аминокислот, содержащих в радикале гидроксильную группу в β-положении по отношению к карбоксильной группе (серин и треонин).
В результате ряда последовательных реакций аминокислота превращается в кетокислоту. Рассмотрим этот процесс на примере превращения треонина в 2-оксобутановую кислоту.

3. Реакции гуанидильной группы.
Гуанидильная группа содержится в радикале аргинина:

Гуанидильная группа аргинина легко отщепляется при гидролизе в избытке гидроксида бария при 1000С с образованием мочевины и орнитина:

Орнитин — α-аминокислота, содержащая в радикале вторую аминогруппу, в состав белков не входит. Появляется в организме в результате гидролитического расщепления аргинина с участием фермента аргиназы. Аргиназа в значительных количествах содержится в печени и в малых количествах в почках и селезенке млекопитающих животных.
Специфические реакции α-аминокислот
Присутствие у одного атома углерода двух функциональных групп (аминогруппы и карбоксильной) приводит к появлению специфических реакций.
1. Образование пептидов — реакция ацилирования одной аминокислоты другой аминокислотой:

Затем дипептид присоединяет следующую молекулу аминокислоты, образуя трипептид, и так далее:

2. Межмолекулярная циклизация — образование дикетопиперазинов.

Реакции аминокислот in vivo
Простые аминокислоты, как и многие другие простые «биологические молекулы», не накапливаются в клетке: как правило, их избыток разрушается при помощи реакций, которые снабжают живую систему энергией. Три основные реакции, катализируемые ферментами, благодаря которым осуществляется превращение аминокислот в клетке, это реакции дезаминирования, переаминирования и декарбоксилирования.
1. Дезаминирование аминокислот
В организме дезаминирование может осуществляться как неокислительным, так и окислительным путём.
Неокислительное дезаминирование встречается, в основном, у бактерий и грибов. Например, превращение аспарагиновой кислоты в фумаровую под действием фермента аспартазы.

Окислительное дезаминирование осуществляется через стадию образования промежуточного имина.
Рассмотрим процесс превращения аланина в пировиноградную кислоту.

Реакции дезаминирования позволяют организму удалять избыток аминокислот, однако при этом повышается концентрация нежелательных азотистых веществ. Высокие концентрации аммиака и его производных токсичны для организма, который поэтому стремится освободиться от них, выделяя лишний азот в виде мочевины или мочевой кислоты.

2. Переаминирование (трансамнирование).
Реакция сводится к взаимопревращению аминогруппы и карбонильной группы под действием ферментов трансаминаз.
Эта реакция служит не только для разрушения аминокислот, но и для их биосинтеза. Рассмотрим реакцию взаимопревращения аспарагиновой кислоты и α-кетоглутаровой в щавелевоуксусную и глутаминовую кислоты:

Эта схема не отражает истинного механизма процесса.
Данное взаимопревращение нуждается в пиридоксальфосфате, который образует имин с исходной аминокислотой, сохраняет аминогруппу при превращении аминокислоты в соответствующую α-кетокислоту и образует имин с другой α-кетокислотой.
Рассмотрим процесс превращения аминокислоты I в α-кетокислоту I и
α-кетокислоты II в аминокислоту II.
Альдегидная группа пиридоксальфосфата образует имин с аминокислотой I, имин далее изомеризуется и после гидролиза выделяет кетокислоту I и пиридоксаминфосфат.

Таким образом, из исходной аминокислоты получилась кетокислота. Образовавшийся пиридоксаминфосфат далее реагирует с другой кетокислотой (кетокислота II), образуя имин, содержащий радикал новой кетокислоты (R¢). Имин далее изомеризуется и после гидролиза образует новую аминокислоту (аминокислота II):

По завершении всей сложной последовательности реакций, после гидролиза пиридоксальфосфат регенерируется и способен принять участие в следующих взаимопревращениях аминокислот и α-кетокислот.
Как своеобразную реакцию взаимопревращения аминокислоты и амидоаминокислоты, сопровождающуюся заменой амидогруппы одной аминокислоты на гидроксильную группу другой, можно рассматривать реакцию взаимодействия L-аспарагиновой кислоты и L-глутамина, катализируемую аспарагинсинтетазой в присутствии АТФ, и приводящую к образованию
L-аспарагина и L-глутаминовой кислоты.

3. Декарбоксилирование аминокислот.

В реакции декарбоксилирования, которая протекает при гниении белков, лизин и орнитин, образуют диамины: кадаверин и путресцин.

Интересной является реакция декарбоксилирования глутаминовой кислоты, так как она приводит к образованию γ-аминомасляной кислоты, которую рассматривают как природный транквилизатор.
Этот процесс также нуждается в присутствии пиридоксальфосфата.

Ярко выраженной биологической активностью обладает амин, образующийся при декарбоксилировании гистидина:

Гистамин является медиатором аллергии: он расширяет все периферические сосуды, что приводит к резкому падению артериального давления, нарушает проницаемость сосудистой стенки, что может быть одной из причин появления отеков, вызывает бронхоспазм и. т.д. Группа препаратов, применяемых в медицине для уменьшения проявления аллергических реакций, так или иначе связанных с гистамином, была названа антигистаминными препаратами.
4. Реакции гидроксилирования и карбоксилирования.
С помощью этих реакций в молекулу органического соединения вводится дополнительная гидроксильная или карбоксильная группы. Реакции протекают при участии соответствующих ферментов и приводят к образованию модифицированных аминокислот. Эти реакции не имеют аналогов в химии in vitro.
Гидроксилированием называют введение в молекулу органического соединения гидроксильной группы. Так, гидроксилирование фенилаланина приводит к образованию тирозина:

Значительный интерес представляет реакция гидроксилирования пролина:

Гидроксилирование пролина необходимо для стабилизации тройной спирали коллагена, которая осуществляется за счет образования водородных связей.
При цинге нарушается гидроксилирование остатков пролина и лизина. В результате образуются менее прочные коллагеновые волокна, что приводит к хрупкости и ломкости кровеносных сосудов.
Карбоксилированием называют введение в молекулу органического соединения карбоксильной группы. Таким образом получают, например,
γ-карбоксиглутаминовую кислоту:

γ-Карбоксиглутаминовая кислота входит в состав белков, участвующих в процессах свертывания крови, так как две близлежащие карбоксильные группы в её структуре способствуют более полному связыванию белковых факторов с ионами кальция:

Нарушение карбоксилирования глутамата приводит к снижению свертываемости крови.
Таким образом, модифицированные аминокислоты, имеющие в своих структурах дополнительные функциональные группы, приобретают свойства, необходимые для выполнения ими специфических функций.
5. Восстановительное аминирование.
Это реакция превращения α-кетокислот в α-аминокислоты осуществляется в организме при участии восстановленной формы никотинамидадениндинуклеотида (НАД∙Н). Так, продуктом метаболизма углеводов является α-кетоглутаровая кислота, которая в результате ряда реакций превращается в глутаминовую кислоту:

6. Альдольное расщепление.
Реакция протекает с α-аминокислотами, содержащими гидроксильную группу в β-положении углеводородного радикала.
Рассмотрим, например, реакцию расщепления серина, в результате которой образуются глицин и формальдегид.

Полипептиды образуются в результате реакции конденсации, протекающей между аминогруппой одной кислоты и карбоксильной группой другой:

Пептиды, содержащие более 10 аминокислот, называют полипептидами. А полипептиды, содержащие более 50 аминокислотных остатков, обычно называют белками. Однако такие градации весьма условны: например, гормон глюкагон, состоящий из 29 аминокислот, называют белковым гормоном. Гормоны окситоцин и вазопрессин содержат всего по 9 аминокислотных остатков.
Поэтому более удачным следует считать различие, проводимое на уровне структуры полимера, более сложном, чем простая аминокислотная последовательность и количественный состав пептида. Полипептиды представляют собой линейные, довольно гибкие молекулы, а длинные цепи белков свернуты в клубок или иную структуру. Многие белки могут иметь в своем составе группы небелкового характера (простетические группы), связанные с полиамидной цепью.
Строение полипептидной цепи и пептидной связи
Мономеры аминокислот, входящие в состав полипептидов, называют аминокислотными остатками. Аминокислотный остаток, имеющий свободную аминогруппу, называют N-концевым и записывают слева пептидной цепи, а имеющий свободную α-карбо-ксильную группу – С-концевым, и записывают справа. Цепь повторяющихся атомов –СН – СО – NH– в полипетидной цепи называется пептидным остовом.
Полипептидная цепь имеет следующий общий вид:

где R1, R2, R3, … Rn – радикалы аминокислот, образующие боковую цепь.
Кислотно-основные свойства пептидов
Многие короткие пептиды были получены в чистом кристаллическом виде. Высокие температуры их плавления указывают на то, что из нейтральных растворов пептиды кристаллизуются в виде диполярных ионов. Поскольку ни одна из
α-карбоксильных групп и ни одна из α-аминогрупп, участвующих в образовании пептидных связей, не может ионизироваться в интервале рН от 0 до 14, кислотно-основные свойства пептидов определяются свободной NH2 группой N-концевого остатка и свободной карбоксильной группой С-концевого остатка пептида и теми
R-группами, которые способны к ионизации. В длинных пептидных цепях число ионизированных
R-групп обычно велико по сравнению с двумя ионизированными группами концевых остатков пептида. Поэтому для характеристики кислотно-основных свойств пептидов мы будем рассматривать короткие пептиды.
Свободная α-аминогруппа и свободная концевая карбоксильная группа в пептидах разделены значительно большим расстоянием, чем в простых аминокислотах, и поэтому электростатические взаимодействия между ними ослаблены. Величины рK для концевых карбоксильных групп в пептидах несколько выше, а для концевых α-аминогрупп несколько ниже, чем в соответствующих свободных аминокислотах. У R-групп в коротких пептидах и в соответствующих свободных аминокислотах величины рK заметно не различаются.
Определение структуры пептидов
Для того чтобы выяснить структуру пептида, необходимо знать следующее:
а) какие аминокислоты входят в состав полипептида;
б) сколько аминокислот каждого вида содержится в пептиде;
в) в какой последовательности эти аминокислоты связаны в цепи.
Для определения состава пептида его подвергают гидролизу в горячей соляной кислоте с С(HCl) = 6 моль/л. Полученную смесь аминокислот анализируют на аминокислотном анализаторе и устанавливают качественный и количественный состав пептида. Зная весовое содержание каждой из полученных аминокислот, можно вычислить количество каждой кислоты и тем самым установить «эмпирическую формулу» пептида, т. е. относительное содержание остатков различных аминокислот в пептиде.
Для вычисления «молекулярной» формулы пептида, то есть для установления действительного числа каждого из остатков в молекуле пептида, необходимо знать его молярную массу, которую определяют различными химическими или физическими методами.
Идентификацию аминокислотных остатков на концах пептидной цепи проводят, используя их отличие от всех остальных звеньев и друг от друга:
N-концевой остаток содержит свободную аминогруппу, а С-концевой остаток содержит свободную карбоксильную группу.
Для идентификации N-концевого остатка используют метод Ф. Сенгера, который основан на реакции свободной аминогруппы пептида с динитрофторбензолом. Реакция протекает по механизму нуклеофильного замещения:

Замещенный пептид подвергают гидролизу, после чего
N-концевой остаток, меченный динитрофенильной группой, выделяют и идентифицируют. N-концевая аминокислота с динитрофторбензолом дает устойчивое, окрашенное в желтый цвет, соединение, которое не разрушается при гидролизе.
Огромный шаг вперед в химии анализа полипептидов был сделан в 1956 году, когда П. Эдман установил, что N-концевую аминокислоту можно удалить при помощи фенилизотиоцианата: (С6Н5 – N = C = S). В результате следующая за ней аминокислота становится N-концевой и её, в свою очередь, также можно удалить, действуя фенилизотиоцианатом. Этот метод определения N-концевых остатков получил название «метод деградации по Эдману».
Наиболее успешным методом определения С-концевых остатков является не химический метод, а ферментативный. Избирательное удаление С-концевого звена осуществляется при помощи фермента карбоксипептидазы, которая расщепляет лишь ту пептидную связь, которая находится в α-положении к свободной
α-карбоксильной группе в полипептидной цепи. Анализ можно повторить на укороченном пептиде, чтобы определить новую С-концевую кислоту.
Однако на практике невозможно определить последовательность остатков аминокислот в длинной пептидной цепи путем ступенчатого удаления концевых остатков. Вместо этого пептид подвергают частичному гидролизу, при котором образуются фрагменты пептидов с укороченной цепью. Эти фрагменты идентифицируют при помощи метода определения концевых групп.
Структура, приписанная пептиду и определенная вышеописанным методом, окончательно подтверждается синтезом этого пептида.


